数据背后的严峻现实
全球范围内,巴德-毕氏综合征(BBS)的发病率约为1/160,000。然而,在部分近亲通婚率较高的封闭人群或特定地域,这一数字可能飙升至1/13,500,飙升近12倍。截至2023年,已发现至少24个(BBS1-BBS24)基因突变可导致BBS,它们共同贡献了约80%的临床病例,这意味着仍有约20%的患者其致病基因尚不明确。这些冷峻的数据揭示了一个事实:BBS并非遥不可及,其诊断高度依赖精准的基因检测技术。
分子核心:纤毛的功能“崩溃”
从分子生物学角度看,BBS是一种典型的“纤毛病”。人体许多细胞表面有一种名为“纤毛”的微小天线状结构。你可以把它想象成细胞的“信号接收站”和“物流调度中心”。BBS相关基因编码的蛋白质,会共同组装成一个叫做“BBSome”的复合体,以及一个“分子伴侣”复合体。
它们的核心工作,就是像“运输车队”一样,沿着纤毛这条“内部公路”,将各种重要的信号蛋白精准运送到指定位置。当BBS基因发生致病突变时,这个运输系统就瘫痪了。 信号蛋白送不到,或者错误蛋白堆积在纤毛里,导致“信号接收站”失灵。这直接影响了细胞感知外界环境、传递发育信号的能力,从而引发多系统病变。

遗传迷宫:为何父母正常孩子却患病?
BBS的遗传模式非常具有迷惑性,这也是临床诊断的难点。它主要遵循常染色体隐性遗传,但存在“三等位基因遗传”等复杂情况。简单说,对于大多数患者,需要从父母那里各继承一个突变的BBS基因副本(即纯合或复合杂合突变)才会发病。
但这里有个关键点:约10%的病例中,患者除了在两个等位基因上携带隐性突变外,还在第三个BBS基因上携带了一个额外的修饰性突变。 这就像给已经故障的系统又加了一根“压垮骆驼的稻草”,显著改变了疾病的严重程度或外显率。因此,仅仅找到一对突变,有时还不能完全解释所有临床表型,需要更全面的基因分析。
检测技术:如何锁定致病元凶?
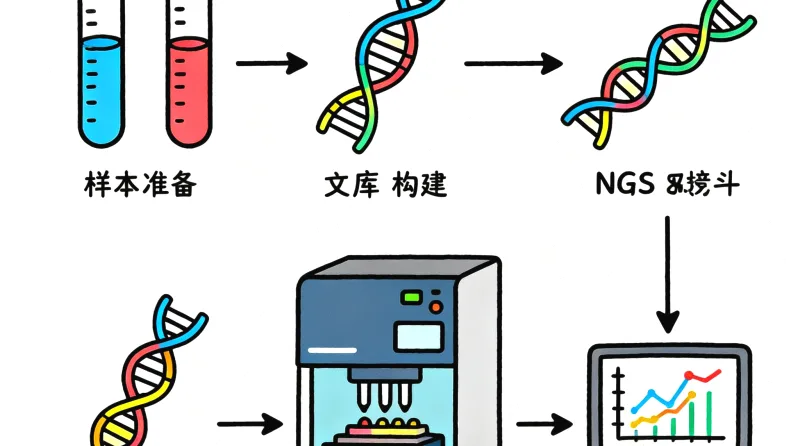
目前,针对BBS的基因检测主要采用高通量测序技术。划重点,全外显子组测序(WES)是首选的策略,因为它能一次性分析所有已知BBS基因及其他相关基因,效率最高。

检测流程步步拆解
在兴宁万核医学检测中心,一份严谨的BBS基因检测报告背后,是标准化的操作流程。
谁需要考虑做这项检测?
基因检测是重要的工具,但并非人人需要。以下人群是主要的适用对象:
理解报告:超越“阳性”与“阴性”
拿到基因检测报告,看懂结论至关重要。报告结果通常分为几类:
说实话,基因检测不是万能的,其核心局限在于对“临床意义未明变异”的解读困境,以及当前技术对某些类型突变(如大片段重复/缺失、三核苷酸重复扩增等)的潜在漏检风险。
要点回顾与未来展望
巴德-毕氏综合征的基因检测,是连接临床表型与分子病因的桥梁。它从分子层面确认诊断,结束“诊断奥德赛”,为患者家庭提供准确的遗传咨询和再发风险评估,并为潜在的未来治疗(如基因治疗)奠定基础。
关键要点总结如下:BBS是高度异质性的纤毛病,遗传模式复杂;全外显子组测序是主流检测方法;检测流程需严格质控;报告解读需谨慎,尤其关注“临床意义未明变异”;阴性结果不能完全排除诊断。 随着基因功能研究和基因编辑技术的进步,今天的精准诊断,正为明天的精准治疗铺平道路。
声明:本站部分信息图片来源于互联网,版权仅归原作者所有,如果侵犯了您的权益,请及时联系我们删除。联系邮箱:824380530@qq.com。发布者:萧晨,转转请注明出处:https://gene.fs371.com/mj5n81kq/